
他身在高楼广厦之中,却有山泽鱼鸟之思

1714 字
9 分钟
PyMOL 自动化批量渲染蛋白复合物界面 (Python 脚本实战)

前言
在进行蛋白-蛋白相互作用(PPI)分析或复合物结构预测后,往往需要对大量的预测结果进行可视化。如果手动在 PyMOL 中逐个加载 PDB 文件并敲击选中、染色、调整透明度的命令,不仅耗时而且极易出错。
本文记录了如何编写一个 Python 脚本,根据 CSV 文件中记录的相互作用界面(Interface)信息,在 PyMOL 中批量、随机抽取目标结构,并自动完成高亮界面的规范化出图。
TIP准备工作
包含 PyMOL 脚本命令的 CSV 数据表,对应的蛋白复合物 PDB 结构库,PyMOL 运行环境
相关核心点: [解决 Interface 搜索] | [精准控制特定链与界面的颜色及透明度]
Interface搜索
import os
import glob
import tempfile
import pandas as pd
import freesasa
from Bio.PDB import MMCIFParser, NeighborSearch, Selection, PDBIO, Select
class ChainSelect(Select):
def __init__(self, allowed_chain_ids):
self.allowed_chain_ids = set(allowed_chain_ids)
def accept_chain(self, chain):
return chain.id in self.allowed_chain_ids
def accept_residue(self, residue):
# 过滤水分子,保留标准氨基酸
if residue.id[0] == 'W':
return 0
return 1
def calc_sasa_for_chains(structure, chain_ids, probe_radius=1.4):
"""使用 FreeSASA 计算指定链集合的 SASA"""
params = freesasa.Parameters({"probe-radius": probe_radius})
io = PDBIO()
io.set_structure(structure)
# 存为临时的 pdb 文件供 FreeSASA 读取
with tempfile.NamedTemporaryFile(suffix=".pdb", delete=False) as tmp:
tmp_path = tmp.name
try:
try:
# 遇到非常规残基时 PDBIO 可能会报警,此处进行保护
io.save(tmp_path, ChainSelect(chain_ids))
except Exception as e:
pass
fs_structure = freesasa.Structure(tmp_path)
result = freesasa.calc(fs_structure, params)
return result.totalArea()
finally:
if os.path.exists(tmp_path):
os.remove(tmp_path)
def analyze_interface(cif_path, chain_a_id='A', chain_b_id='B', distance=5.0, probe_radius=1.4):
parser = MMCIFParser(QUIET=True)
try:
structure = parser.get_structure('complex', cif_path)
except Exception as e:
return {"error": f"读取文件失败: {e}"}
model = structure[0]
try:
chain_a = model[chain_a_id]
chain_b = model[chain_b_id]
except KeyError as e:
return {"error": f"找不到指定的链 {e}"}
# 1) 距离法提取界面残基用于 PyMOL
atoms_a = Selection.unfold_entities(chain_a, 'A')
atoms_b = Selection.unfold_entities(chain_b, 'A')
ns_a = NeighborSearch(atoms_a)
ns_b = NeighborSearch(atoms_b)
interface_a = set()
interface_b = set()
for atom_a in atoms_a:
if ns_b.search(atom_a.coord, distance):
interface_a.add(atom_a.get_parent().id[1])
for atom_b in atoms_b:
if ns_a.search(atom_b.coord, distance):
interface_b.add(atom_b.get_parent().id[1])
seq_a = "+".join(str(res) for res in sorted(interface_a))
seq_b = "+".join(str(res) for res in sorted(interface_b))
pymol_cmd = (
f"select interface_A, chain {chain_a_id} and resi {seq_a}; "
f"select interface_B, chain {chain_b_id} and resi {seq_b}; "
f"select interface_all, interface_A or interface_B"
)
# 2) BSA = SASA(A) + SASA(B) - SASA(complex)
try:
sasa_a = calc_sasa_for_chains(structure, [chain_a_id], probe_radius=probe_radius)
sasa_b = calc_sasa_for_chains(structure, [chain_b_id], probe_radius=probe_radius)
sasa_complex = calc_sasa_for_chains(structure, [chain_a_id, chain_b_id], probe_radius=probe_radius)
bsa = sasa_a + sasa_b - sasa_complex
except Exception as e:
return {"error": f"SASA/BSA 计算失败: {e}"}
# 3) 过滤标准 (BSA > 400 且 双方参与残基数 >= 10)
pass_bsa = bsa > 400.0
pass_residue_count = (len(interface_a) >= 10) and (len(interface_b) >= 10)
pass_all = pass_bsa and pass_residue_count
folder_name = os.path.basename(os.path.dirname(cif_path))
return {
"file_name": os.path.basename(cif_path),
"folder": folder_name,
"chain_A": chain_a_id,
"chain_B": chain_b_id,
"distance_threshold": distance,
"probe_radius": probe_radius,
"interface_A_residue_count": len(interface_a),
"interface_B_residue_count": len(interface_b),
"sasa_A": round(sasa_a, 3),
"sasa_B": round(sasa_b, 3),
"sasa_complex": round(sasa_complex, 3),
"bsa": round(bsa, 3),
"pass_bsa_gt_400": pass_bsa,
"pass_interface_residue_ge_10": pass_residue_count,
"pass_all_filters": pass_all,
"pymol_script": pymol_cmd,
}
def batch_process_interfaces(base_dir, output_csv, max_files=None, distance=5.0, probe_radius=1.4):
results = []
cif_files = glob.glob(os.path.join(base_dir, "**/*.cif"), recursive=True)
print(f"找到 {len(cif_files)} 个 CIF 文件...")
if max_files is not None:
cif_files = cif_files[:max_files]
print(f"测试模式开启:仅处理前 {max_files} 个文件")
else:
print("开始处理所有文件...")
for idx, cif_path in enumerate(cif_files, start=1):
folder_name = os.path.basename(os.path.dirname(cif_path))
parts = folder_name.split('_')
# 尝试从文件夹名称中推断链 ID,默认为 A 和 B
chain_a, chain_b = 'A', 'B'
if len(parts) >= 3 and parts[-1].isalpha() and len(parts[-1]) == 2:
chain_a, chain_b = parts[-1][0], parts[-1][1]
print(f"[{idx}/{len(cif_files)}] 处理: {folder_name}")
res = analyze_interface(
cif_path,
chain_a_id=chain_a,
chain_b_id=chain_b,
distance=distance,
probe_radius=probe_radius
)
if "error" in res:
print(f" -> 失败: {res['error']}")
results.append({
"file_name": os.path.basename(cif_path),
"folder": folder_name,
"error": res["error"],
})
else:
print(f" -> 成功: 计算得 BSA = {res['bsa']} Ų")
results.append(res)
df = pd.DataFrame(results)
# 确保 pymol_script 放在 CSV 文件的最后一列,方便复制
if not df.empty and "pymol_script" in df.columns:
cols = df.columns.tolist()
cols.remove("pymol_script")
cols.append("pymol_script")
df = df[cols]
df.to_csv(output_csv, index=False, encoding='utf-8-sig')
print(f"\n处理完成,共记录 {len(results)} 条数据,已保存到: {output_csv}")
# ==========================================
# 批量处理运行代码
# ==========================================
predictions_dir = xx
output_csv_path = "interface_metrics_clean_bsa_test.csv"
# 先小规模测试:仅处理前 5 个
batch_process_interfaces(
predictions_dir,
output_csv_path,
max_files=300,
distance=5.0,
probe_radius=1.4
)
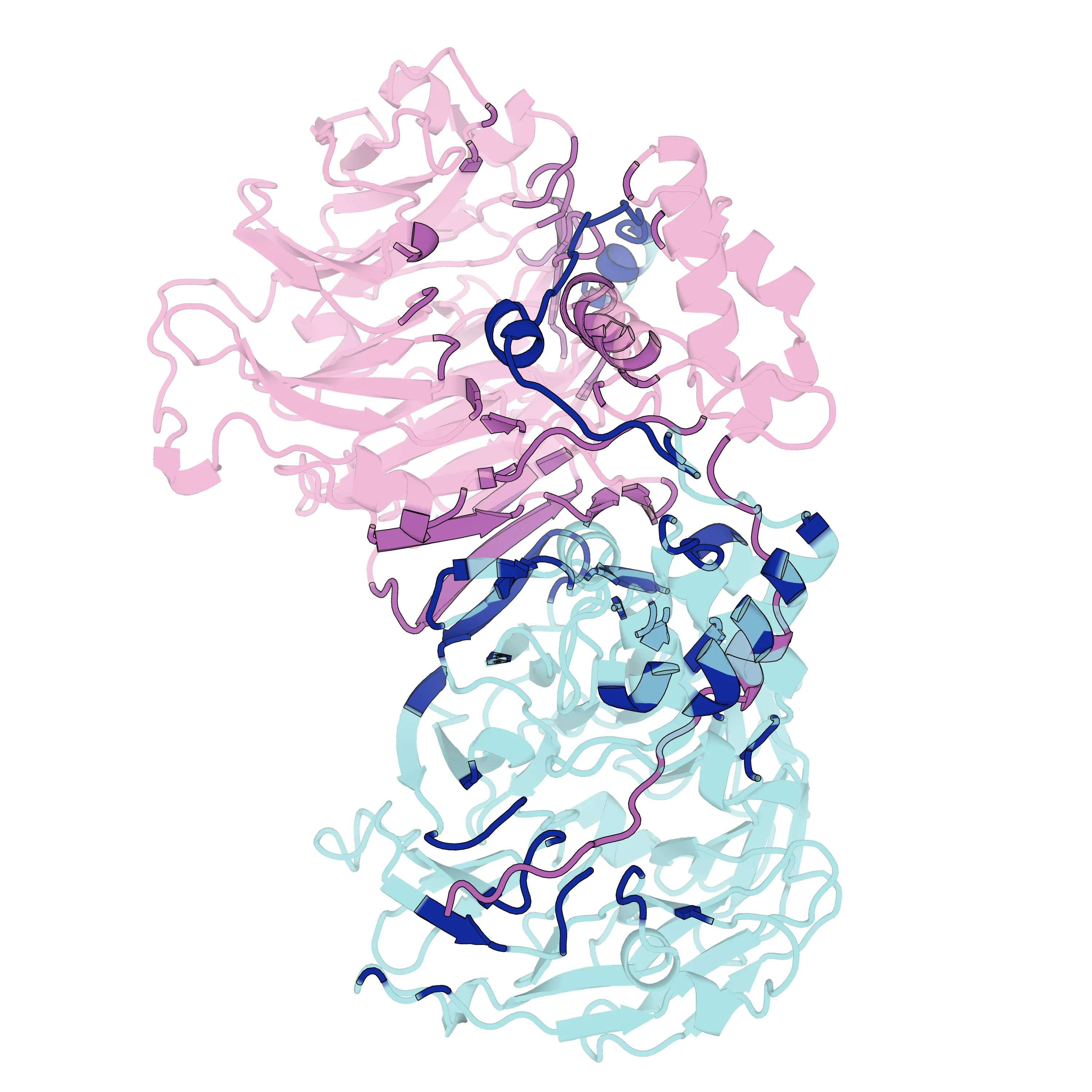
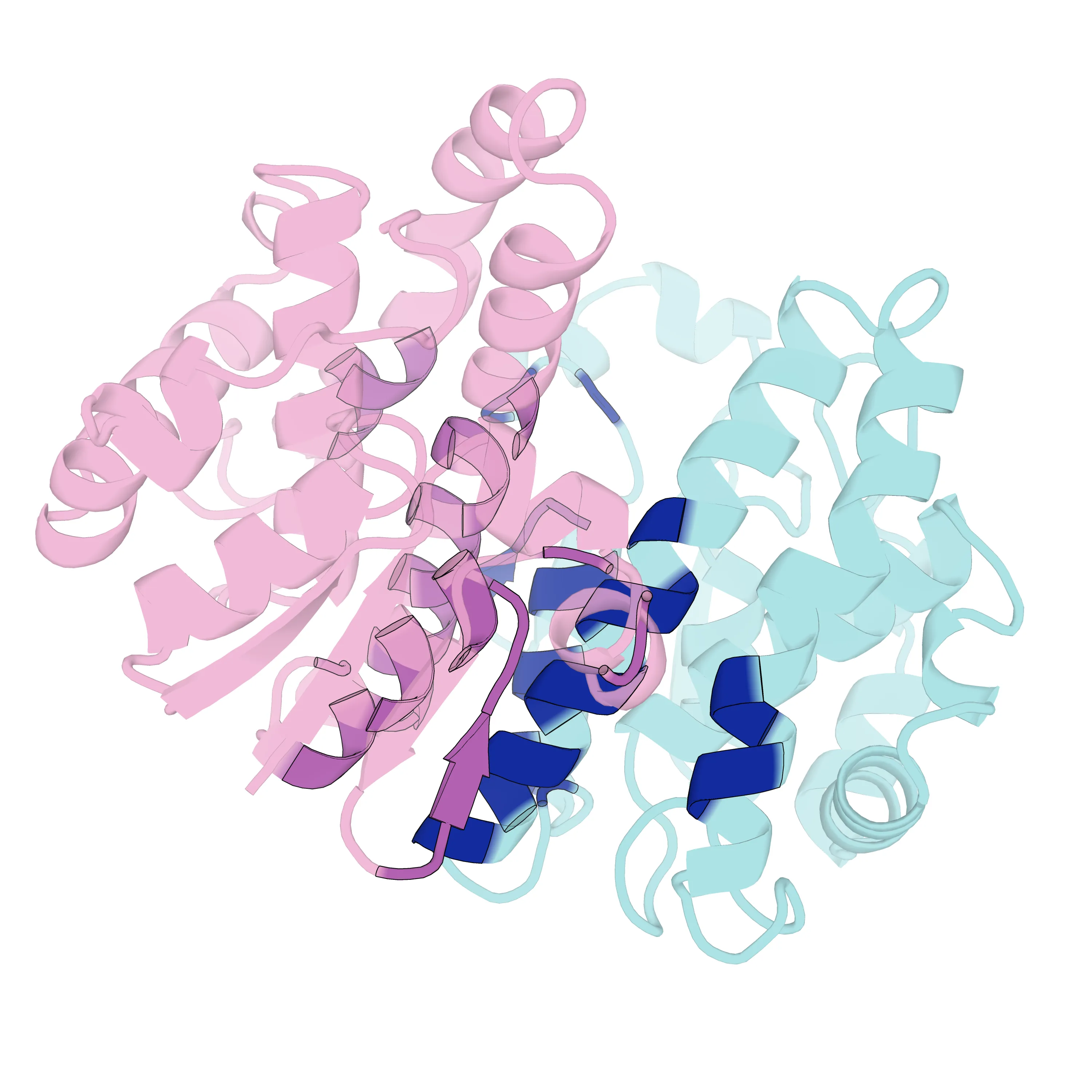
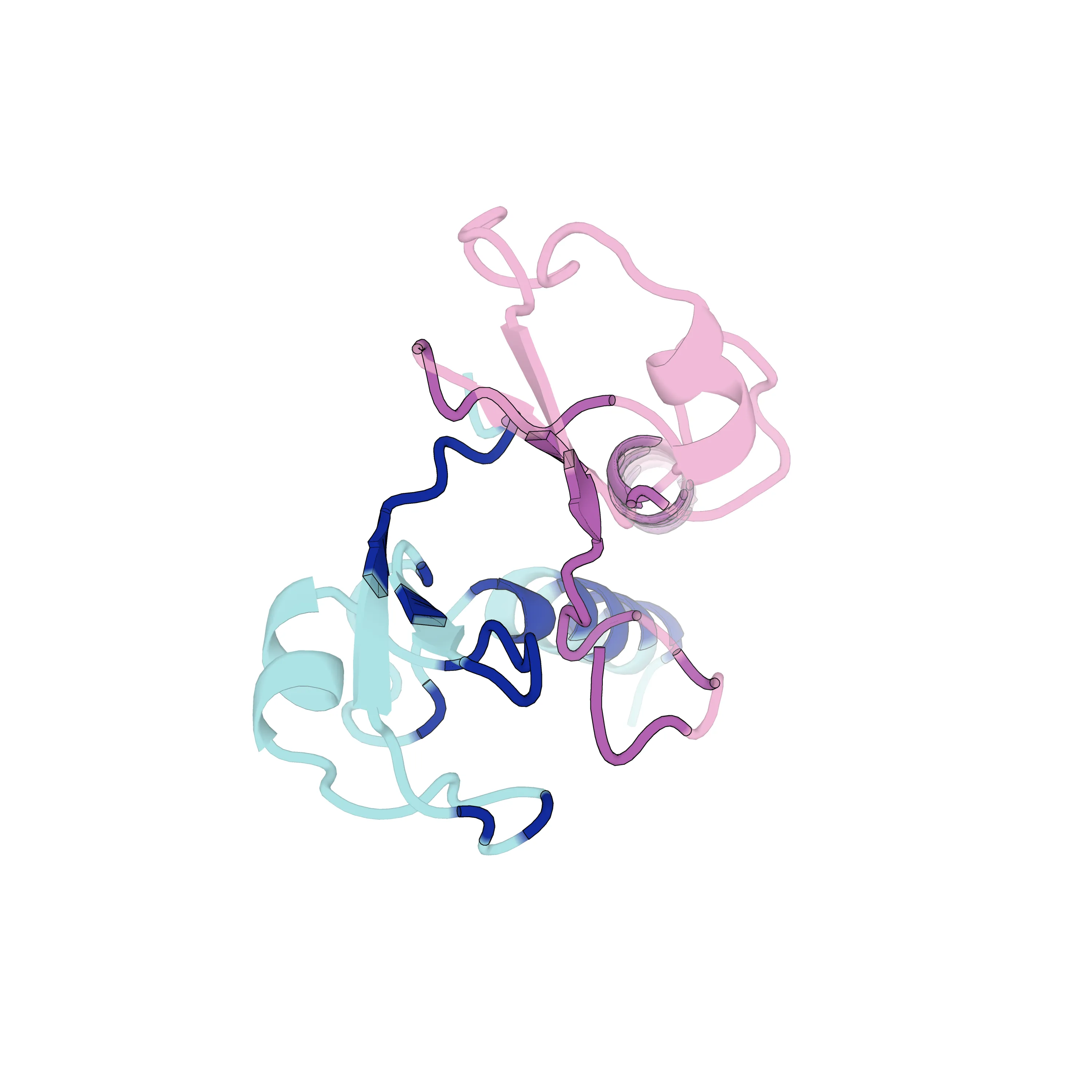
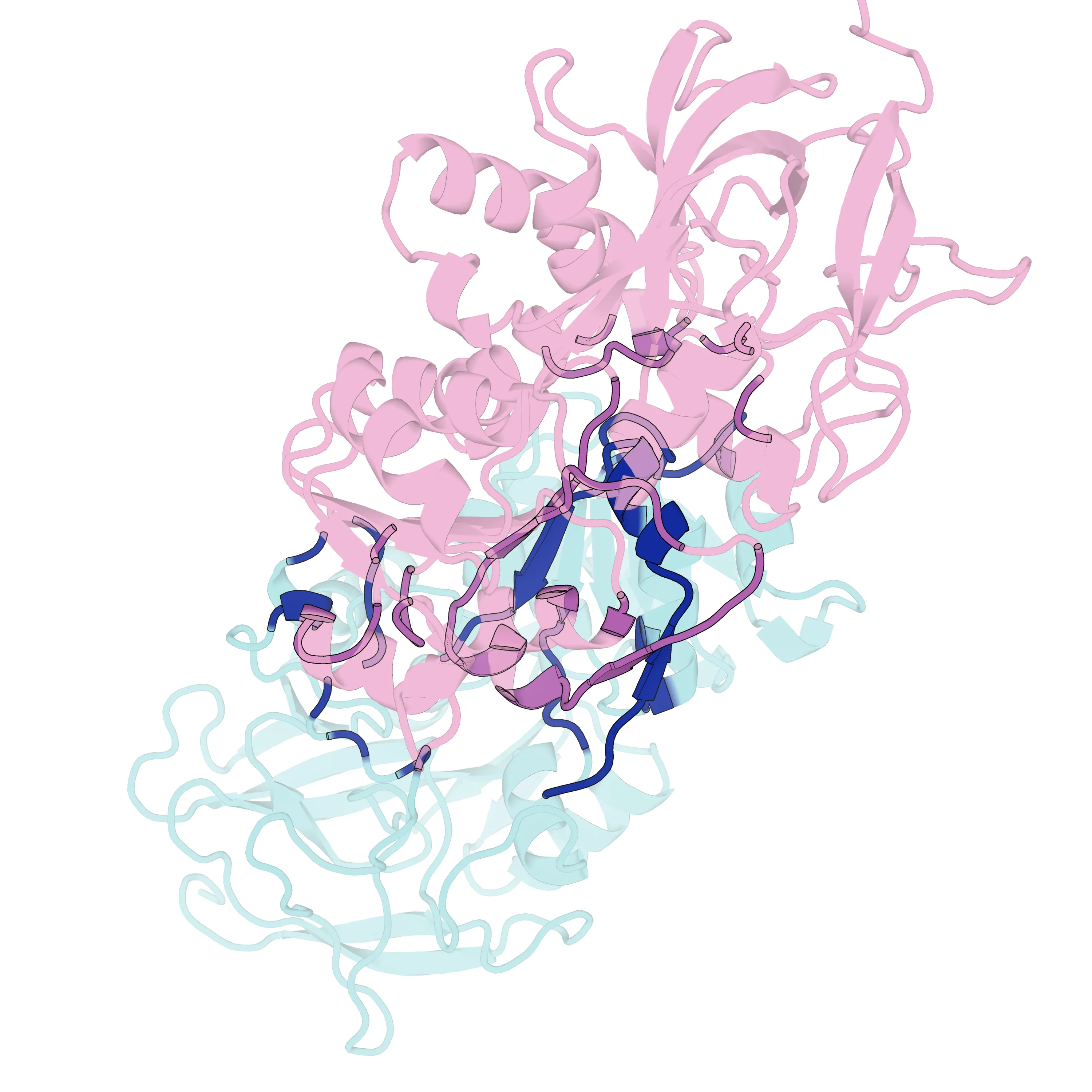
渲染逻辑优化:颜色与透明度控制
为了让相互作用的界面在图片中足够醒目,我们采用了以下渲染策略:
- 隐藏 Sticks:整体统一使用 Cartoon 卡通模型展示。
- 基础染色:Chain A 染成
pink,Chain B 染成cyan。 - 高亮 Interface:Chain A 的界面氨基酸染成更深的
magenta,Chain B 的界面染成blue。 - 透明度剥离:将非 Interface 区域的透明度设置为 20%(
0.2),而 Interface 保持完全不透明(0.0),以此突出核心作用区域。
完整自动化脚本
以下是经过优化并加入“随机抽取 + 子集导出”功能的完整 Python 脚本:
import csv
import os
import random
import re
import time
import glob
from pymol import cmd
def _safe_name(text):
return re.sub(r"[^0-9A-Za-z_]+", "_", text)
def _infer_pdb_id(file_name):
if not file_name:
return "UNK0"
m = re.match(r"^([A-Za-z0-9]{4})", file_name)
return m.group(1).upper() if m else "UNK0"
def _parse_pymol_script(script):
if not script:
return None
m_a = re.search(r"select\s+interface_A\s*,\s*chain\s+([A-Za-z0-9])\s+and\s+resi\s+([^;]+)", script, re.IGNORECASE)
m_b = re.search(r"select\s+interface_B\s*,\s*chain\s+([A-Za-z0-9])\s+and\s+resi\s+([^;]+)", script, re.IGNORECASE)
if not (m_a and m_b):
return None
return m_a.group(1), m_a.group(2).strip(), m_b.group(1), m_b.group(2).strip()
def _choose_loaded_object(cmd, row):
loaded = set(cmd.get_names("objects"))
file_name = row.get("file_name", "")
folder = row.get("folder", "")
pdb_id = _infer_pdb_id(file_name)
candidates = [f"prot_{pdb_id}", folder, os.path.splitext(file_name)[0] if file_name else "", pdb_id]
for c in candidates:
if c and c in loaded:
return c
return None
def _resolve_structure_path(row, structures_root_dir):
file_name = row.get("file_name", "")
folder = row.get("folder", "")
if structures_root_dir and folder and file_name:
p = os.path.join(structures_root_dir, folder, file_name)
if os.path.exists(p): return p
if structures_root_dir and file_name:
p = os.path.join(structures_root_dir, file_name)
if os.path.exists(p): return p
if structures_root_dir and file_name:
matches = glob.glob(os.path.join(structures_root_dir, "**", file_name), recursive=True)
if matches: return matches[0]
return None
def _get_or_load_object(cmd, row, structures_root_dir):
obj_name = _choose_loaded_object(cmd, row)
if obj_name: return obj_name, None
structure_path = _resolve_structure_path(row, structures_root_dir)
if not structure_path:
return None, "未找到对应结构文件"
file_name = row.get("file_name", "")
folder = row.get("folder", "")
pdb_id = _infer_pdb_id(file_name)
load_name = _safe_name(f"prot_{pdb_id}_{folder or os.path.splitext(file_name)[0]}")
try:
cmd.load(structure_path, load_name)
# 强制检查 PyMOL 中是否真正创建了该对象
if load_name not in cmd.get_names("objects"):
return None, f"PyMOL 解析文件失败 (可能文件为空或缺少后缀): {structure_path}"
return load_name, None
except Exception as e:
return None, f"加载结构发生 Python 异常: {e}"
def run_random20_from_csv(
csv_path,
structures_root_dir,
output_dir,
n=20,
seed=20260313,
command_log_path=None,
width=3000,
height=3000,
dpi=300,
):
if command_log_path is None:
command_log_path = os.path.join(output_dir, "random20_pymol_commands.txt")
if not os.path.exists(output_dir):
os.makedirs(output_dir)
with open(csv_path, "r", encoding="utf-8-sig", newline="") as f:
reader = csv.DictReader(f)
fieldnames = reader.fieldnames
rows = list(reader)
valid_rows = [r for r in rows if (r.get("pymol_script") or "").strip()]
if not valid_rows:
raise ValueError("CSV 中没有可用的 pymol_script 列内容。")
random.seed(seed)
sample_size = min(n, len(valid_rows))
picked = random.sample(valid_rows, sample_size)
# 导出随机选中的子集供后续核对
out_csv_path = os.path.join(output_dir, "random20_selected.csv")
with open(out_csv_path, "w", encoding="utf-8-sig", newline="") as f:
writer = csv.DictWriter(f, fieldnames=fieldnames)
writer.writeheader()
writer.writerows(picked)
cmd.bg_color("white")
cmd.set("ray_opaque_background", "on")
log_lines = [
f"CSV: {csv_path}",
f"Selected Sub-CSV: {out_csv_path}",
f"Sample size: {sample_size} (Random seed: {seed})",
"=" * 80
]
for i, row in enumerate(picked, start=1):
file_name = row.get("file_name", "")
folder = row.get("folder", "")
script = row.get("pymol_script", "")
obj_name, load_err = _get_or_load_object(cmd, row, structures_root_dir)
if not obj_name:
log_lines.append(f"[{i}] SKIP {file_name} | 原因: {load_err}")
continue
parsed = _parse_pymol_script(script)
if not parsed:
log_lines.append(f"[{i}] SKIP {file_name} | 原因: 无法解析 pymol_script")
continue
chain_a, resi_a, chain_b, resi_b = parsed
tag = _safe_name(folder or file_name or f"item_{i}")
sel_a = f"interface_A_{tag}"
sel_b = f"interface_B_{tag}"
sel_all = f"interface_all_{tag}"
cmd.select(sel_a, f"{obj_name} and chain {chain_a} and resi {resi_a}")
cmd.select(sel_b, f"{obj_name} and chain {chain_b} and resi {resi_b}")
cmd.select(sel_all, f"{sel_a} or {sel_b}")
# 出图风格设置
cmd.disable("all")
cmd.enable(obj_name)
cmd.show("cartoon", obj_name)
# 染色
cmd.color("pink", f"{obj_name} and chain {chain_a}")
cmd.color("cyan", f"{obj_name} and chain {chain_b}")
cmd.color("magenta", sel_a)
cmd.color("blue", sel_b)
# 透明度设置
cmd.set("cartoon_transparency", 0.2, f"{obj_name} and not {sel_all}")
cmd.set("cartoon_transparency", 0.0, sel_all)
cmd.zoom(obj_name)
pdb_id = _infer_pdb_id(file_name)
out_png = os.path.join(output_dir, f"{i:02d}_{pdb_id}_{tag}.png")
print(f"[{i}/{sample_size}] 渲染: {out_png}")
cmd.png(out_png, width=width, height=height, dpi=dpi, ray=1)
time.sleep(0.5)
log_lines.extend([
f"[{i}] file={file_name} object={obj_name}",
f"select {sel_a}, {obj_name} and chain {chain_a} and resi {resi_a}",
f"select {sel_b}, {obj_name} and chain {chain_b} and resi {resi_b}",
f"select {sel_all}, {sel_a} or {sel_b}",
"-"
])
with open(command_log_path, "w", encoding="utf-8") as f:
f.write("\n".join(log_lines))
在 PyMOL 的命令行中,可以通过如下包含嵌套 Python 块的 .pml 脚本一键调用执行:
run C:/Users/86173/Desktop/MPU_phd/20260310Biodata/pymol_random20_from_csv.py
python
run_random20_from_csv(
csv_path=r"C:\Users\86173\xx\interface_metrics_clean_bsa_test.csv",
structures_root_dir=r"C:\Users\86173\xx\complex_predictions_pack.tar\complex_predictions_pack\complex_predictions\predictions",
output_dir=r"C:\Users\86173\xx\interface",
n=20,
seed=20260313
)
python end
渲染效果展示
通过上述脚本,最终渲染导出了指定的高清图片及封面:
图片参考 蛋白复合物 Interface 渲染效果
PyMOL 自动化批量渲染蛋白复合物界面 (Python 脚本实战)
https://sereinna.github.io/posts/蛋白蛋白interface绘制/