
他身在高楼广厦之中,却有山泽鱼鸟之思

1014 字
5 分钟
weight_picture
前言
TIP准备工作 分子
smiles\
如何对分子进行模型权重图绘制
from rdkit.Chem import AllChem as Chem
from rdkit.Chem import Draw
from rdkit.DataStructs import ConvertToNumpyArray
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
%matplotlib inline
import skchem
from ipywidgets import interact, Dropdown, Text, Box
from matplotlib.colors import ListedColormap
def sym_twod_gaussian(x, mu=np.zeros((2,))):
return np.exp(-(x - mu).dot(x - mu))
def perpendicular(a) :
b = np.empty_like(a)
b[0] = -a[1]
b[1] = a[0]
return b
def highlight(m, weights=None, bond_gap=0.05, padding=0.1, vis_mode='circle', cmap='Blues'):
two_d_idx = Chem.Compute2DCoords(m)
conf = m.conformers[two_d_idx]
coords = np.array(conf.atom_positions)
boundary = (coords.min(axis=0) - padding, coords.max(axis=0) + padding)
length = boundary[0] - boundary[1]
aspect = length[0] / length[1]
tall = aspect < 1
if tall:
width, height = 12, 12 * aspect
else:
width, height = 12 * aspect, 12
plt.figure(figsize=(width, height), facecolor=None, dpi=300)
plt.xlim(boundary[0][0], boundary[1][0])
plt.ylim(boundary[0][1], boundary[1][1])
if vis_mode == 'circle':
weights = 4000 * weights if weights is not None else 0
plt.scatter(coords[:, 0], coords[:, 1], s=weights, facecolors=None)
elif vis_mode == 'contour':
weights = weights if weights is not None else 0
x, y = coords[:,0], coords[:,1]
xx, yy = np.mgrid[boundary[0][0]:boundary[1][0]:50j, boundary[0][1]:boundary[1][1]:50j]
positions = np.vstack([xx.ravel(), yy.ravel()])
values = np.vstack([x, y])
kernel = lambda x: sum(w * sym_twod_gaussian(x, c) for w, c in zip(weights, coords[:,:2]))
f = np.reshape(np.array([kernel(i) for i in positions.T]).T, xx.shape)
def my_cmp(cmap):
cmap = plt.get_cmap(cmap)
new_cmap = cmap(np.linspace(0, 1, 256))
new_cmap[:1, :] = np.array([1,1,1,1])
return ListedColormap(new_cmap)
plt.contourf(xx, yy, f, cmap=my_cmp(cmap))
else:
raise NotImplementedError
# 绘制原子标签
for i, a in enumerate(m.atoms):
hs = a.GetNumImplicitHs() + a.GetNumExplicitHs()
if hs > 1:
label = '${}H_{}$'.format(a.element, hs)
elif hs == 1:
label = '${}H$'.format(a.element)
else:
label = '$' + a.element + '$'
plt.text(coords[i, 0] - 0.1, coords[i, 1], label, verticalalignment='center', fontsize=12)
# 绘制键
for b in m.bonds:
idxs = b.GetBeginAtomIdx(), b.GetEndAtomIdx()
coord1, coord2 = coords[idxs[0], :2], coords[idxs[1], :2]
norm = np.linalg.norm(coord2 - coord1)
vect = (coord1 - coord2) / norm
coord1, coord2 = coord1 - 0.1 * vect, coord2 + 0.1 * vect
# 绘制单键
if b.order == 1:
plt.plot([coord1[0], coord2[0]], [coord1[1], coord2[1]], c='black')
# 绘制芳香键
if b.order == 1.5:
plt.plot([coord1[0] + 0.5 * bond_gap * perpendicular(vect)[0], coord2[0] + 0.5 * bond_gap * perpendicular(vect)[0]],
[coord1[1] + 0.5 * bond_gap * perpendicular(vect)[1], coord2[1] + 0.5 * bond_gap * perpendicular(vect)[1]], '--', c='black')
plt.plot([coord1[0] - 0.5 * bond_gap * perpendicular(vect)[0], coord2[0] - 0.5 * bond_gap * perpendicular(vect)[0]],
[coord1[1] - 0.5 * bond_gap * perpendicular(vect)[1], coord2[1] - 0.5 * bond_gap * perpendicular(vect)[1]], c='black')
# 绘制双键
if b.order == 2:
plt.plot([coord1[0] + 0.5 * bond_gap * perpendicular(vect)[0], coord2[0] + 0.5 * bond_gap * perpendicular(vect)[0]],
[coord1[1] + 0.5 * bond_gap * perpendicular(vect)[1], coord2[1] + 0.5 * bond_gap * perpendicular(vect)[1]], c='black')
plt.plot([coord1[0] - 0.5 * bond_gap * perpendicular(vect)[0], coord2[0] - 0.5 * bond_gap * perpendicular(vect)[0]],
[coord1[1] - 0.5 * bond_gap * perpendicular(vect)[1], coord2[1] - 0.5 * bond_gap * perpendicular(vect)[1]], c='black')
# 绘制三键
if b.order == 3:
plt.plot([coord1[0], coord2[0]], [coord1[1], coord2[1]], c='black')
plt.plot([coord1[0] + bond_gap * perpendicular(vect)[0], coord2[0] + bond_gap * perpendicular(vect)[0]],
[coord1[1] + bond_gap * perpendicular(vect)[1], coord2[1] + bond_gap * perpendicular(vect)[1]], c='black')
plt.plot([coord1[0] - bond_gap * perpendicular(vect)[0], coord2[0] - bond_gap * perpendicular(vect)[0]],
[coord1[1] - bond_gap * perpendicular(vect)[1], coord2[1] - bond_gap * perpendicular(vect)[1]], c='black')
plt.axis('off')
return plt.gca()
# 绘制分子图
def plot_mol(smiles, atom_weights, vis_mode='contour', cmap='Blues', save_path='plot_mol.png'):
m = skchem.Mol.from_smiles(smiles) # 从 SMILES 创建分子对象
print(len(atom_weights))
print(len(m.GetAtoms()))
assert len(atom_weights) == len(m.GetAtoms()) # 确保原子数和权重数匹配
highlight(m, weights=atom_weights, padding=1.5, vis_mode=vis_mode, cmap=cmap) # 绘制分子
plt.savefig(save_path, bbox_inches='tight', transparent=True) # 保存图片
plt.show() # 显示图片
plt.close() # 关闭图形
# 示例:随机生成 7 个原子权重,绘制分子图
weights1 = np.random.rand(26)
weights2 = np.random.rand(17)
weights3 = np.random.rand(19)
weights4 = np.random.rand(19)
#plot_mol(smiles='CN[C@@H](C)C(=O)N[C@H](C(=O)N1CC2=CC(OCCOCCOCCOCCOCCOC(=O)N3CCC[C@@H](N4N=C(C5=CC=C(OC6=CC=C(F)C=C6F)C=C5)C(C(N)=O)=C4N)C3)=CC=C2C[C@H]1C(=O)N[C@@H]1CCCC2=CC=CC=C21)C(C)(C)C', atom_weights=weights) # 绘制苯酚分子图
smiles_list = [
'COc1ccc2c(c1)OCCN(CC2)C(=O)CCCCN3CCOCC3',
'CCN(CC)CCCC(=O)Nc1ccccc1',
'CN1CCN(CC1)CCCC(=O)Nc2ccccc2',
'COc1cccc2c1C(=O)NC(C2)c3ccccc3'
]
plot_mol(smiles='COc1ccc2c(c1)OCCN(CC2)C(=O)CCCCN3CCOCC3', atom_weights=weights1) #
plot_mol(smiles='CCN(CC)CCCC(=O)Nc1ccccc1', atom_weights=weights2) #
plot_mol(smiles='CN1CCN(CC1)CCCC(=O)Nc2ccccc2', atom_weights=weights3) #
plot_mol(smiles='COc1cccc2c1C(=O)NC(C2)c3ccccc3', atom_weights=weights4) #
如何对分子属性权重图绘制
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.Draw import SimilarityMaps
from matplotlib.colors import LinearSegmentedColormap
from rdkit.Chem.Draw import rdMolDraw2D
# 定义多个分子的 SMILES
smiles_list = [
'COc1ccc2c(c1)OCCN(CC2)C(=O)CCCCN3CCOCC3',
'CCN(CC)CCCC(=O)Nc1ccccc1',
'CN1CCN(CC1)CCCC(=O)Nc2ccccc2',
'COc1cccc2c1C(=O)NC(C2)c3ccccc3'
]
# 参数:调整权重范围的缩放因子
scaling_factor = 1 # 强化权重的显著性
# 设置高权重和低权重的颜色
low_weight_color = '#6595e8' # 低权重颜色
high_weight_color = '#d7697e' # 高权重颜色
custom_cmap = LinearSegmentedColormap.from_list('custom', [low_weight_color, "white", high_weight_color])
# 绘制多个分子的权重图并保存为 SVG
for idx, smiles in enumerate(smiles_list):
# 生成分子对象
mol = Chem.MolFromSmiles(smiles)
if mol is None:
print(f"Invalid SMILES: {smiles}")
continue
# 计算 Gasteiger 电荷
AllChem.ComputeGasteigerCharges(mol)
contribs = [float(mol.GetAtomWithIdx(i).GetProp('_GasteigerCharge')) for i in range(mol.GetNumAtoms())]
# **直接限制贡献范围**:
# 通过调整映射范围增强高权重区域染色
max_contrib = max(contribs)
min_contrib = min(contribs)
midpoint = (max_contrib + min_contrib) / 3.5 # 中间点
scaled_contribs = [
((c - midpoint) * scaling_factor) / (max_contrib - min_contrib + 1e-5) for c in contribs
]
# 使用 RDKit 的 GetSimilarityMapFromWeights 绘制分子权重图
fig = SimilarityMaps.GetSimilarityMapFromWeights(
mol, scaled_contribs, colorMap=custom_cmap, contourLines=10
)
# 使用 RDKit 的 MolDraw2DSVG 绘制并保存为 SVG
drawer = rdMolDraw2D.MolDraw2DSVG(500, 500) # 定义 SVG 绘图区域
drawer.SetDrawOptions(drawer.drawOptions())
drawer.DrawMolecule(mol, highlightAtoms=list(range(mol.GetNumAtoms())))
drawer.FinishDrawing()
# 保存 SVG 文件
svg_file = f"molecule_{idx + 1}.svg"
with open(svg_file, 'w') as f:
f.write(drawer.GetDrawingText())
print(f"SVG saved: {svg_file}")
# 提示保存完成
print("All molecules have been processed and saved as SVG files.")
作图结果示例
图片参考 属性图 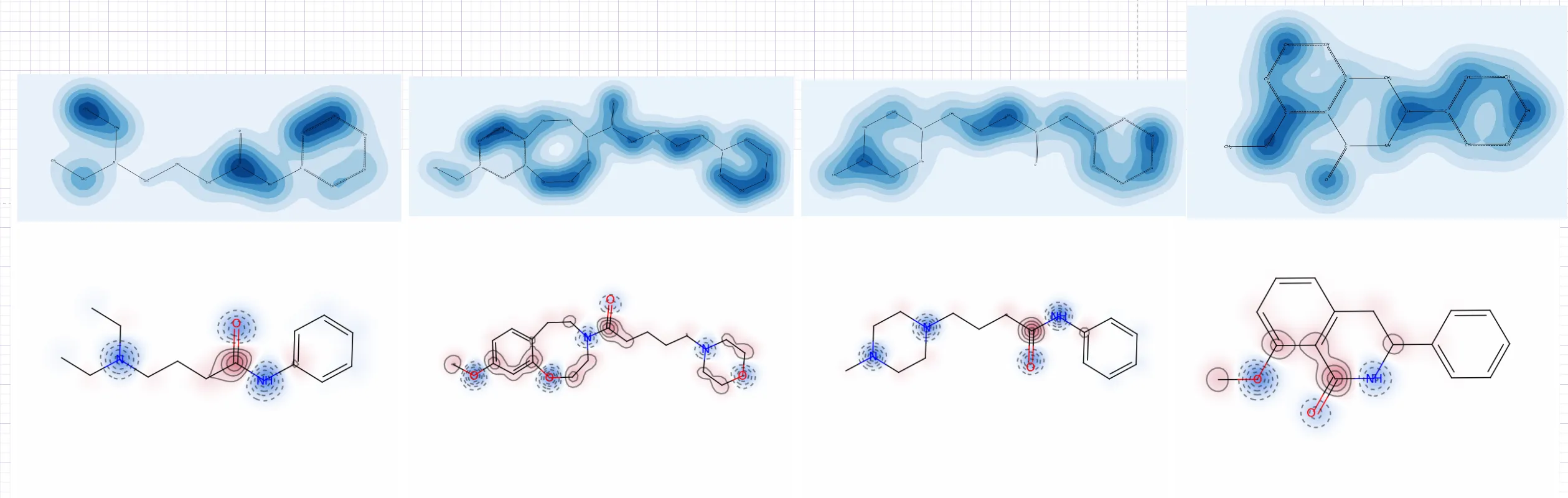
weight_picture
https://sereinna.github.io/posts/weight_pocture/