前言
TIP准备工作 文献阅读器
zetoro, 相关文献摘要
研究当前的暴力破解或人工智能支持的 uHTVS 方法是否可以构成 DEL 筛查的计算替代方案
介绍
通常,将适当修饰的蛋白质靶标固定在固体支持物(例如链霉亲和素珠)上,然后与完整的 DEL 文库一起孵育。通过一系列的清洗步骤,大多数文库成员被从溶液中去除,只留下那些对蛋白质靶标具有高亲和力的成员,然后通过适当的洗脱程序将其回收。然后使用 PCR 解码这些命中化合物的 DNA 标签扩增和高通量 DNA 测序,然后通常通过脱 DNA 合成和/或正交 DNA 分析来验证最佳命中。 从我们在对实验高通量筛选 (HTS) 和 VS 的方法学比较开始,并通过使用不同大小的文库(从 105 到 108 种化合物)进行 DEL 筛选的三个案例研究向上发展
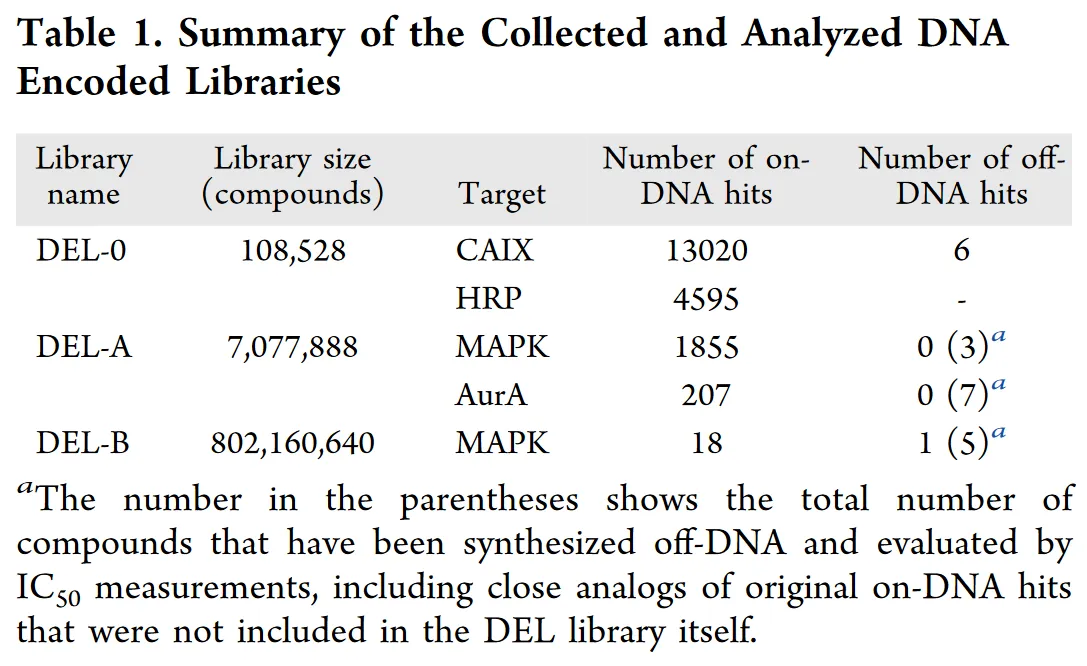
三个数据库的内容总结
Enumeration and Preprocessing of the Libraries
DEL-0 数据集
- 来源:从 Gerry 等人的研究中提取(参考文献 35)。
- 数据获取:从相关的补充材料中获取标记为 DEL 的化合物的 SMILES 字符串,并使用 Schrödinger 的 LigPrep 在 pH 7.4 下进行预处理。
- 化合物数量:数据集由 8 个骨架、114 个位于位置 1 的构建块(BB1)和 119 个位于位置 2 的构建块(BB2)组成,共计 108,528 个化合物。
- 唯一化合物:实际有 107,616 个唯一化合物,因为 BB2 中的 70 和 77 是相同的片段。但实验测量对这些条目显示了不同的值,因此我们分别处理了重复的化合物,尽管它们的对接评分相同。
- 目标蛋白结合评估:针对碳酸酐酶 IX(CAIX)和辣根过氧化物酶(HRP),通过归一化倍数(Fn)进行评估。
- 命中化合物分类:具有 Fn 大于或等于 1 的化合物被分别视为对 CAIX 或 HRP 的 On-DNA 命中物。
- Off-DNA 命中物:从出版物中收集,根据组成的构建块识别合成的化合物。
DEL-A 数据集
- 来源:从 Clark 等人的出版物中提取(参考文献 30)。
- 组成:由三个构建块组成,每个有 192 个不同的分子,总计 7,077,888 个化合物。
- 枚举方法:由于缺乏化合物标识符,使用 Marvin 软件(参考文献 37)进行枚举。
- 将构建块导入 MarvinSketch(参考文献 38),移除保护基,设置连接原子,根据构建块位置构建 R-基团。
- 使用 Marvin 的 cxcalc(参考文献 39)的 Markush 枚举批处理模式创建完整 DEL-A 库化合物的 SMILES 字符串。
- 预处理:使用 LigPrep(参考文献 40)对枚举的化合物进行预处理,设置与 DEL 数据集描述的类似。
- 命中化合物:具有实验读取计数值的 DEL-A 数据集化合物被视为命中物。
- 命中识别:由于相关出版物的支持材料仅包含命中化合物的构建块信息,命中识别是通过重建原始化合物,然后使用 RDKit 软件包(参考文献 41)和 KNIME 平台(参考文献 42)进行识别。
DEL-B 数据集
- 来源:也从 Clark 等人的工作中提取(参考文献 30)。
- 组成:每个化合物由四个构建块组成,分别是 Fmoc 保护的胺、双功能酸和两个不同组的胺,成员数分别为 192、32、340 和 384 个片段。
- 化合物数量:这四个构建块构成了一个包含 802,160,640 个化合物的 DEL 库。
- 枚举方法:使用与 DEL-A 部分描述的相同方法,使用 Marvin 软件进行枚举。
- 预处理:由于 DEL-B 库中的化合物数量庞大,分子的预处理方式与 DEL-0 和 DEL-A 数据集不同。
- 命中化合物:具有实验读取计数值的 DEL-B 数据集化合物被视为命中物。
- 命中识别:命中识别方式与 DEL-A 部分描述的相同(见表 1)。
内容
超高通量虚拟筛选(uHTVS)的进展
随着计算能力的提高,虚拟筛选也逐渐成为探索化学空间的一种有效手段。文中提到的uHTVS工具包括GPU加速的AutoDock、深度学习模型驱动的Deep Docking、以及用于形状筛选的GPU版ROCS算法等。这些工具使得筛选数亿种化合物成为可能。
虚拟筛选的挑战:尽管uHTVS技术不断进步,但其计算成本依然很高,尤其是在筛选数据集较大的情况下。为了解决这一问题,近年来开发了几种基于逻辑层的工作流,如V-SYNTHES(通过分子片段组合进行筛选)和Deep Docking(用深度学习模型预测对接评分)。
虚拟筛选与DNA编码库的对比
作者分析了虚拟筛选与DNA编码库在方法论上的异同点,指出两者各有优劣。虽然虚拟筛选在筛选速度和灵活性上具有优势,但DELs在处理大规模化合物库方面仍然表现出色。
三个案例研究
DEL-0数据集:包含10万多个化合物,筛选目标为过氧化物酶(HRP)和碳酸酐酶IX(CAIX)。通过对这些化合物进行虚拟筛选,作者比较了不同对接算法(如AutoDockGPU、Glide)在恢复DEL筛选命中率方面的表现。
DEL-A数据集:包含700多万个化合物,针对Aurora A激酶(AurA)和MAPK蛋白的筛选。该数据集规模较大,作者使用了Glide HTVS对其进行对接,并通过Deep Docking流程进行AI辅助筛选。
DEL-B数据集:包含8亿多种化合物,是最大的筛选数据集。由于数据量过大,无法进行暴力对接,作者仅使用了AI辅助工作流进行筛选,重点分析了Deep Docking的效果。
性能评估:作者通过接收者操作特性(ROC)曲线和富集因子(EF)评估了虚拟筛选的效果,结果表明,虽然Deep Docking能够有效筛选化合物,但其性能依赖于对接算法的质量。
- DEL-0 数据集 数据集简介:DEL-0 数据集是一个相对较小的DNA编码库,包含107,616种独特化合物。其分子结构由8种不同的骨架、114种N-端修饰元素以及118种Suzuki反应派生的元素组合而成。目标蛋白是过氧化物酶(HRP)和碳酸酐酶IX(CAIX),这两者都与生物催化、癌症治疗及细胞调控等相关。
筛选方法:在对DEL-0数据集进行筛选时,研究人员使用了不同的对接算法,包括:
Glide(SP单精度、HTVS高通量虚拟筛选、SPcon带位置约束):这是一个商用的对接软件。 AutoDockGPU:一个开源的GPU加速对接软件。对CAIX的对接还采用了AutoDock4Zn,这是一种为锌金属蛋白优化的力场。 结果与分析:
对接性能:通过接收者操作特性(ROC)曲线评估不同对接方法的表现,Glide系列方法总体表现优于AutoDockGPU,尤其是在对CAIX和HRP进行对接时表现更好。然而,AutoDockGPU因其开源特性,依然是替代商用软件的一个重要选择。 时间要求:Glide的计算时间明显更长,因为它依赖CPU计算,而AutoDockGPU利用GPU加速。因此,在多核CPU环境下,Glide的HTVS模式在时间效率上表现更优异。 结果差异:有趣的是,虚拟筛选的对接评分与实验命中率之间没有显著的相关性。这表明某些构件在虚拟筛选中被高估,反映了虚拟筛选算法在处理DEL数据时存在噪声问题。 总的来说,DEL-0数据集的案例展示了uHTVS与DEL筛选相结合的潜力,但也揭示了虚拟筛选在噪声处理和命中率预测方面的挑战。
- DEL-A 数据集 数据集简介:DEL-A 数据集包含7,077,888种化合物,目标蛋白包括Aurora A激酶(AurA)和p38丝裂原活化蛋白激酶(MAPK)。AurA在细胞分裂、肿瘤干细胞性和细胞迁移中起到关键作用,而MAPK则调控细胞对压力的反应,参与肿瘤发生等多种生物过程。
筛选方法:由于DEL-A数据集的规模较大,研究人员主要采用了Glide HTVS模式,并引入了Deep Docking(深度对接)这一AI辅助筛选流程。Deep Docking使用深度学习模型预测对接评分,从而减少需要进行实际对接的化合物数量。
结果与分析:
暴力对接与AI辅助筛选的比较: 使用Glide HTVS对整个DEL-A数据集进行暴力对接得出的ROC曲线表明,虽然对AurA的筛选表现良好(部分曲线达到了预期的命中率),但对MAPK的筛选表现较差。尤其是在前1,000个评分最高的化合物中,MAPK几乎没有命中。 Deep Docking结合了Glide HTVS和AutoDockGPU的深度学习模型,用更少的化合物进行了对接。对于AurA,AI辅助筛选恢复了84.1%的实验命中,但对于MAPK的恢复率较低,仅为27.1%。 AI模型的性能差异:Deep Docking的性能依赖于所用的对接引擎。在使用AutoDockGPU时,很多化合物未能成功对接,这影响了ML模型的表现,导致命中率较低。相反,Glide HTVS的筛选结果更好,特别是对AurA的命中率较高。 该案例展示了AI辅助工作流的潜力,同时也表明,在更大规模的化合物库筛选中,暴力对接虽然耗时,但仍具有优势,特别是在AI模型预测不够准确时。
- DEL-B 数据集 数据集简介:DEL-B数据集是最大的一个案例,包含802,160,640种化合物,目标蛋白为MAPK。由于该数据集的规模庞大,研究人员无法进行暴力对接,而是完全依赖于AI辅助筛选工作流(Deep Docking与Glide HTVS结合)。
筛选方法:与DEL-A相似,研究人员使用Deep Docking对DEL-B数据集进行筛选。该流程从库中随机抽取1.6%的化合物进行训练,然后利用深度学习模型预测剩余化合物的对接评分。
结果与分析:
命中率恢复:尽管Deep Docking标记了1.8百万个潜在命中化合物,但实际恢复的实验命中率仅为77.8%。尽管筛选出了部分符合预期的化合物,但许多命中化合物在筛选过程中未被成功识别。 性能评价:与随机抽样进行的比较表明,Deep Docking工作流在MAPK的筛选上具有一定优势,尤其是在前1%的化合物中,富集因子(EF)达到了11.1。 该案例展示了AI辅助筛选在处理超大规模化合物库中的重要性,尽管其结果未能完全恢复所有实验命中,但其高效性使其在面对规模极大的化学空间时表现出潜力。
- 构件分析 构件贡献的评估:在所有三个案例中,研究人员还对化合物的分子构件进行了分析,评估了不同构件在命中化合物中的重要性。通过计算构件在不同筛选命中化合物中的Pbind值,作者发现实验结果和虚拟筛选结果之间的差异较大。例如,某些在实验筛选中被认为重要的构件,在虚拟筛选中并未显示出相应的重要性。
构件分析表明,虚拟筛选结果中一些构件的重要性可能被夸大或低估,表明当前虚拟筛选算法在捕捉化学构件贡献方面仍存在不足。